| Version 3 (modified by , 14 years ago) (diff) |
|---|
Table of Contents
SNP calling pipeline
Status: Alpha
Authors: Freerk van Dijk, Morris Swertz
Based on Broad pipeline.
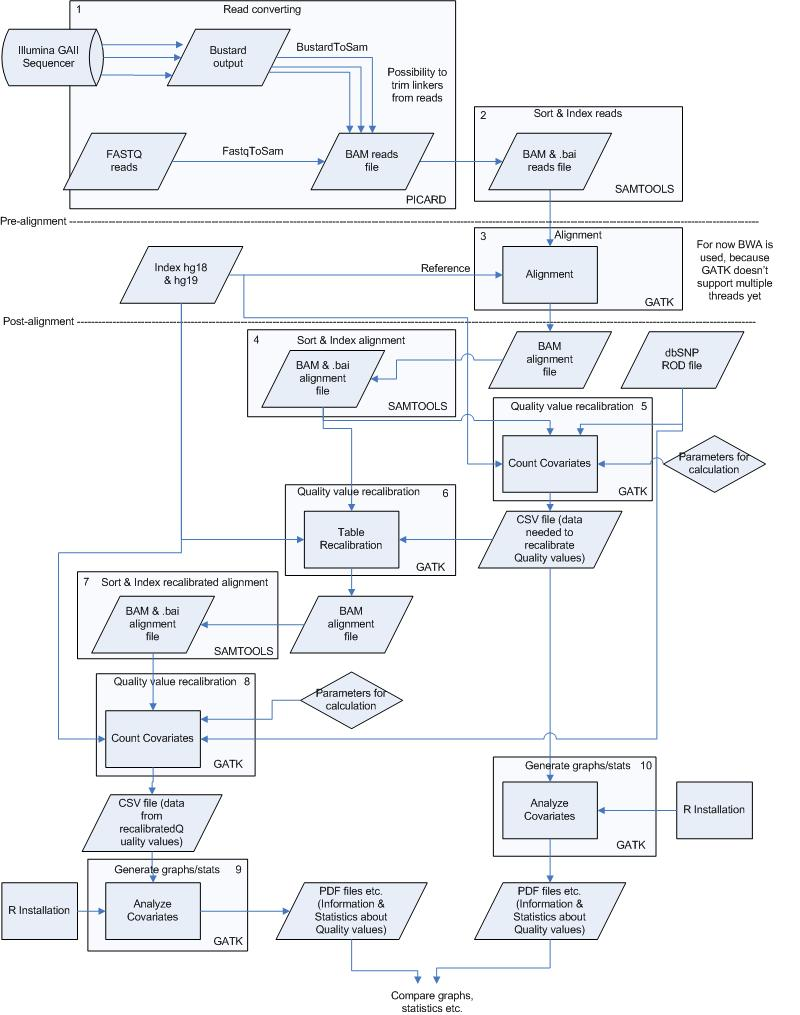
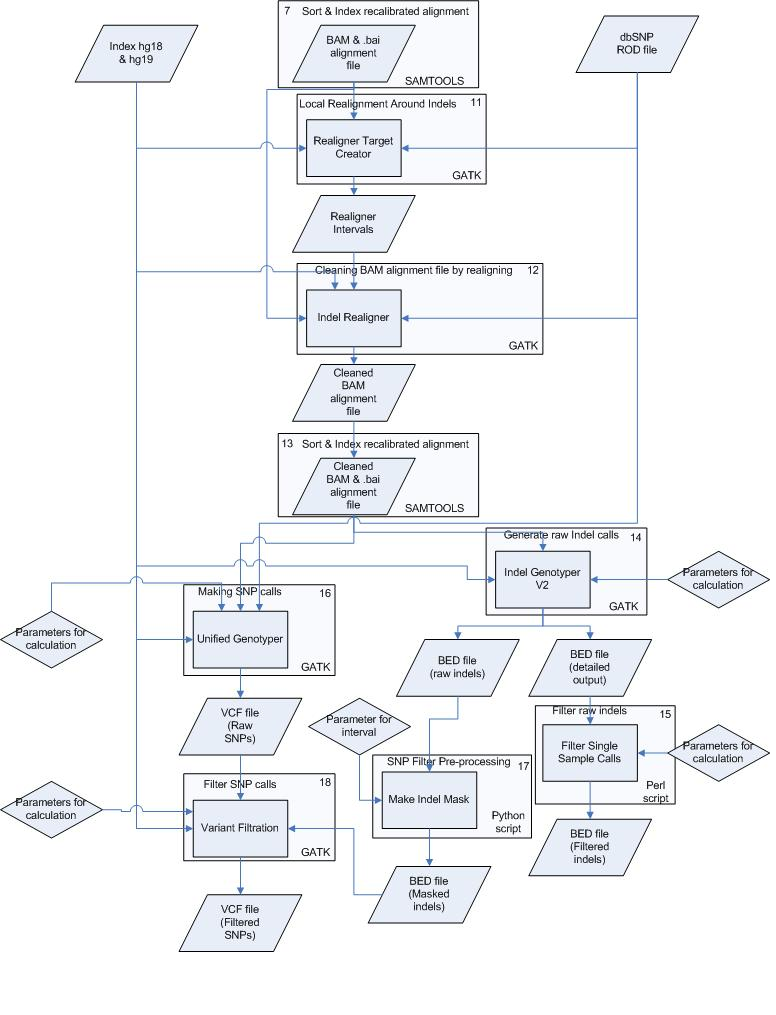
To perform the analysis as fast and good as possible the pipeline has been divided into several small processes. These processes are all numbered and can be found in figure 1 & 2 in the appendix. The processes including commands, input and output files are described below, starting with pre-alignment.
A. Pre-alignment
This process involves all needed steps to prepare the raw sequencing reads for an alignment using BWA. These steps are all conducted using PICARD. An overview of the steps can be found in Appendix A.
1) Read converting
This step involves converting the output from the Illumina GAII, FASTQ format, to binary SAM format (BAM). To do this FastqToSam.jar can be used. The following command was used:
java -jar !FastqToSam.jar FASTQ=../testrun/s_2_sequence041090.txt QUALITY_FORMAT=Illumina OUTPUT=../testrun/s_2_sequence.bam SAMPLE_NAME=s_2_test_sample PLATFORM_UNIT=barcode_13434HWUSIsomething PLATFORM=illumina SORT_ORDER=coordinate
It is also possible to convert raw BUSTARD data to BAM using BustardToSam.jar. The advantage of this tool is the multiple lane input, in theory all lanes from one flow cell can be converted parallel. To remove possible linkers from reads a tool named ExtractIlluminaBarcodes.jar can be used. Since there isn’t any data including these barcodes this and the BustardToSam haven’t been tested yet.
2) Sort & index reads
The output of FastqToSam.jar needs to be sorted and indexed in order to use it as input for the alignment. To sort these files SAMtools was used. The following commands can be executed on the BAM file:
./samtools sort ../testrun/s_2_sequence041090_aligned.bam ../testrun/s_2_aligned_sorted
The postfix .bam is automatically added to the output file.
The corresponding BAM index file can be created using:
./samtools index ../testrun/s_2_aligned_sorted.bam ../testrun/s_2_aligned_sorted.bam.bai
B. Alignment
This consists of alignment using BWA, coverting steps and the sorting and indexing of the output BAM file.
3) Alignment]
To align the reads to a reference genome the GATK uses BWA. The indices used for this step were already made for an earlier Galaxy project. GATK uses an own version of BWA which doesn’t support multiple threading yet. To overcome this problem alignment was performed using the standard BWA. The following command was executed:
./bwa aln -l 101 -k 4 -t 16 ../Sting/dist/resources/index_hg36.fa ../testrun/s_2_sequence041090.txt -f ../testrun/s_2_sequence041090_aligned.sai
This results in an alignment file in .sai format. To convert this to SAM format the following command was used:
./bwa aln -l 101 -k 4 -t 16 ../Sting/dist/resources/index_hg36.fa ../testrun/s_2_sequence041090.txt -f ../testrun/s_2_sequence041090_aligned.sai
To convert this SAM file to BAM SAMtools was used, the command:
./bwa aln -l 101 -k 4 -t 16 ../Sting/dist/resources/index_hg36.fa ../testrun/s_2_sequence041090.txt -f ../testrun/s_2_sequence041090_aligned.sai
4) Sort & index alignment
After this converting step the BAM file needs to be sorted & indexed again using the following command:
./samtools sort ../testrun/s_2_sequence041090_aligned.bam ../testrun/s_2_aligned_sorted
The index was created using:
./samtools index ../testrun/s_2_aligned_sorted.bam ../testrun/s_2_aligned_sorted.bam.bai
NOTE. Keep track of the reference build used. UCSC uses human build hg18 and hg19, which correspond to build 36 and 37 of the NCBI. These builds contains the same information and sequences but different headers etc. The dbSNP ROD files which need to be used further on in the pipeline can be downloaded from UCSC. To overcome any compatibility problems it is advised to use hg18 and hg19 as reference during alignment and further calculations.
C. Post-alignment
This step involves all processes executed on the generated alignments. These steps include quality value recalibration, SNP/indel calling, annotation and the calculation of statistics as well.
5) Quality value recalibration (Count covariates)]
To determine the covariates influencing base quality scores in the BAM file obtained after alignment this step is performed. This results in a CSV file. The following command was used to execute this step:
java -jar GenomeAnalysisTK.jar -R resources/index_hg36.fa --default_platform illumina --DBSNP resources/hg36/snp130.txt -I ../../testrun/s_2_aligned_sorted.bam --max_reads_at_locus 20000 -T CountCovariates -cov ReadGroupcovariate -cov QualityScoreCovariate -cov CycleCovariate -cov DinucCovariate -recalFile ../../testrun/recal_data.csv
The parameters used for this calculation need to be tested more. This to see the influence of each parameter on the output results.
6) Quality value recalibration (Table recalibration)
This step includes the actual quality value recalibration. The recalibration takes place using the generated CSV file as input. The output of this process is the alignment file with recalibrated quality values in BAM format. This step was executed using the following command:
java -jar GenomeAnalysisTK.jar -R resources/index_hg36.fa --default_platform illumina -I ../../testrun/s_2_aligned_sorted.bam -T TableRecalibration -outputBam ../../testrun/s_2_aligned_recalibrated.bam -recalFile ../../testrun/recal_data.csv
7) Sort & index recalibrated alignment]
The resulting BAM file needs to be sorted and indexed again. This step needs to be repeated every time an analysis or calculation has been performed on a BAM file. To sort and index this file use the commands explained in step 4.
8) Quality value recalibration (Count covariates)
This step is executed on the obtained BAM file after the quality value recalibration. This file is used as input, the command used further is exactly the same as the command used in step 5.
9) Generate graphs/stats
To generate several statistics and graphs on the recalibrated alignment the AnalyzeCovariates.jar tool can be used. This tool uses the generated .CSV file as an input. The output is a directory containing several statistics and graphs about the quality values etc. The following command was used:
Java –jar AnalyzeCovariates.jar –recalFile ../../testrun/recal_data.csv –outputDir ../../testrun/analyzecovariates_output –resources resources/index_hg36.fa –ignoreQ 5
10) Generate graphs/stats
This step calculates the same as step 9, but used the CSV file from step 5 as input. The output from this step can be compared to the output of step 9. By comparing the output from both above mentioned steps the increase and changes in quality value etc. can be visualized.
D. SNP & indel calling
This part of the pipeline involves the tools for SNP and indel calling. This part also includes local realignment and filtering for potential SNPs. The following steps divide this part in sub processes.
11) Local realignment around indels
The first step is to determine small suspicious intervals in the alignment. These intervals will then be realigned to get an optimal output result. To do this, these intervals were calculated using the following command:
java -jar GenomeAnalysisTK.jar -T RealignerTargetCreator -I ../../testrun/s_2_aligned_recalibrated_sorted.bam -R resources/index_hg36.fa -o ../../testrun/forRealigner.intervals -D resources/hg36/snp130.txt
This process outputs a file containing intervals on which realignment should be executed.
12) Cleaning BAM alignment file by realignment
For each detect interval a local realignment takes place. This results in a cleaned BAM file on which SNP and indel calling takes place. The local alignment was performed using the following command:
java -Djava.io.tmpdir=/tmp -jar GenomeAnalysisTK.jar -I ../../testrun/s_2_aligned_recalibrated_sorted.bam -R resources/index_hg36.fa -T IndelRealigner -targetIntervals ../../testrun/forRealigner.intervals --output ../../testrun/s_2_aligned_cleaned.bam -D resources/hg36/snp130.txt
13) Sort & index cleaned alignment === The cleaned BAM file is again sorted and indexed using the command as described in step 4.
14) Generate raw indel calls
In this step raw indel calls are made. This output two bed files, the first containing raw indels, the second detailed output. This process was executed using the following command:
Java –jar GenomeAnalysisTK.jar –T IndelGenotyperV2 –R resources/index_hg36.fa --I ../../testrun/s_2_aligned_cleaned.bam –O ../../testrun/indels.raw.bed –o ../../testrun/detailed.output.bed –verbose –mnr 1000000
The generated raw indel file will later be used in step 17.
15) Filter raw indels
To retrieve the actual list of indels this last step needs to be performed. This results in a filtered list containing all the detected indels. The following command (Perl script) was used:
perl ../perl/filterSingleSampleCalls.pl --calls ../../testrun/detailed.output.bed --max_cons_av_mm 3.0 --max_cons_nqs_av_mm 0.5 --mode ANNOTATE > ../../testrun/indels.filtered.bed
16) Making SNP calls
To make SNP calls this process needs to be executed. This results in a VCF file containing raw SNPs. This list will be filtered in step 18, using output from step 17. To generate this list of raw SNPs the following command was used:
Java –jar GenomeAnalysisTK.jar –R resources/index_hg36.fa –T UnifiedGenotyper -I ../../testrun/s_2_aligned_cleaned.bam -D resources/hg36/snp130.txt –varout ../../testrun/s2_snps.raw.vcf –stand_call_conf 30.0
17) SNP filter pre-processing
In this step the raw indel file created in step 14 was used. Based on the regions described in this file the tool filters out SNPs located near these potential indels. The output of this file is used for filtering the SNP calls in step 18. The following command was used:
Python ../python/makeIndelMask.py ../../testrun/indels.raw.bed 10 ../../testrun/indels.mask.bed
The number in this command stands for the number of bases that will be included on either side of the indel.
18) Filter SNP calls
The last step is the filtering on the list of SNPs. This filtering step includes a list of parameters which haven’t been tested yet. These parameters aren’t included in this command because they have to be tested. The following command was executed:
Java –jar GenomeAnalysisTK.jar –T VariantFiltration –R resources/index_hg36.fa –O ../../testrun/s2_snps.filtered.vcf –B ../../testrun/s2_snps.raw.vcf –B ../../testrun/indels.mask.bed –maskName InDel –clusterWindowSize 10 –filterExpression “AB > 0.75 && DP > 40
| DP > 100 | MQ0 > 40 | SB > -0.10” –filterName “StandardFilters” –filterExpression “MQ0 >= 4 && ((1.0 * DP) > 0.1)” –filterName “HARD_TO_VALIDATE” |
The output of this step is the list of SNPs found in this individual. More information on the parameters used can be found on: http://www.broadinstitute.org/gsa/wiki/index.php/VariantFiltrationWalker & http://www.broadinstitute.org/gsa/wiki/index.php/Using_JEXL_expressions
E. Calculating quality metrics & removing duplicate reads
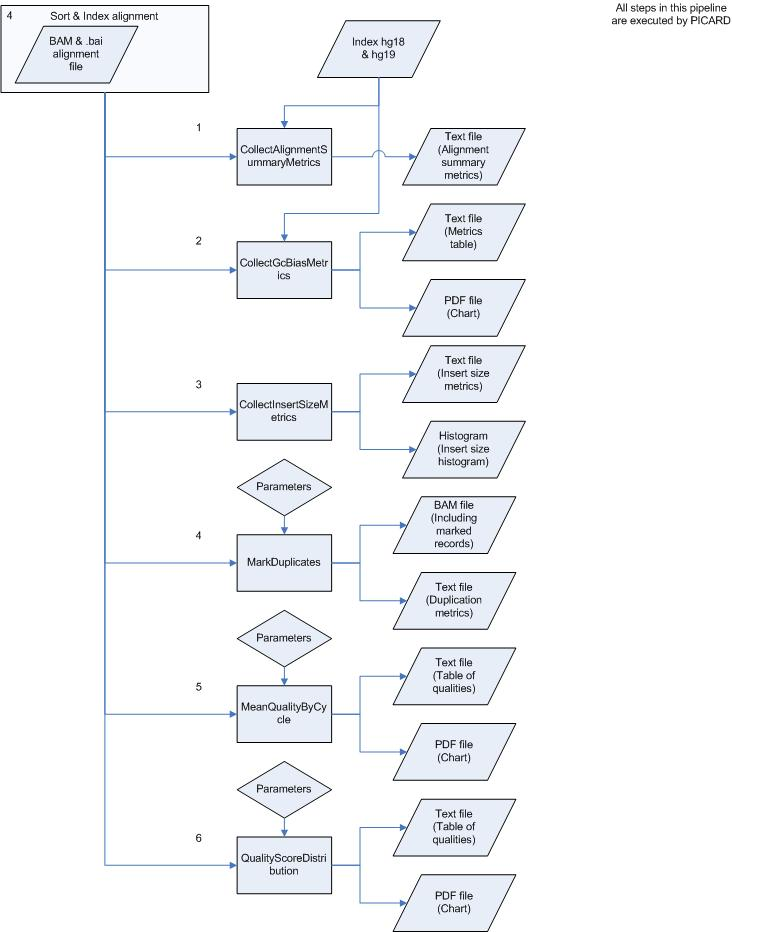
To remove duplicate reads after alignment PICARD can be used. This tool can also calculate different metrics on alignment data. Therefore PICARD was installed and tested on an aligned and sorted BAM file. A process overview can be seen in figure 3. This process starts at step 4 of the analysis pipeline and uses the sorted alignment file as input. A complete testrun also was performed on this tool and developed pipeline. The command, results etc. can be found in Complete_Picard_test_run.doc.
Remarks & Suggestions
There are several other tools which can be implemented into the pipeline, one of these tools is an implementation for Beagle. This tool is only usable in an experimental way at the moment. The only use cases supported by this interface are:
- Inferring missing genotype data from call sets (e.g. for lack of coverage in low-pass data)
- Genotype inference for unrelated individuals.
The basic workflow for this interface is as follows:
- After variants are called and possibly filtered, the GATK walker ProduceBeagleInputWalker will take the resulting VCF as input, and will produce a likelihood file in BEAGLE format.
- User needs to run BEAGLE with this likelihood file specified as input.
- After Beagle runs, user must unzip resulting output files (.gprobs, .phased) containing posterior genotype probabilities and phased haplotypes.
- User can then run GATK walker BeagleOutputToVCFWalker to produce a new VCF with updated data. The new VCF will contain updated genotypes as well as updated annotations.
GATK also has the ability to run process in parallel. This option is only supported by a limited amount of tools. It can be imported in every tool using parallelism, more information on this can be found on: http://www.broadinstitute.org/gsa/wiki/index.php/Parallelism_and_the_GATK
Manuals, help etc
More information about the GATK can be found at:
http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit
A forum containing a lot of background information can be found at:
http://getsatisfaction.com/gsa
Information on the other tools used can be found at;
SAMtools: http://samtools.sourceforge.net/
PICARD: http://picard.sourceforge.net/index.shtml
Another source of valuable information on anything NGS related:
http://seqanswers.com/forums/index.php
= [ Appendix A – Schematic overview of the pipeline] =
Figure 1. Overview of the analysis pipeline created using the GATK. The steps/processes are all numbered. More information on these steps can be found in the corresponding text sessions.
Figure 2. Overview of the analysis pipeline created using the GATK. This part starts at step 7 from figure 1. The steps/processes are all numbered. More information on these steps can be found in the corresponding text sessions.
Figure 3. Overview of the PICARD pipeline. This tool performs several quality checks after the alignment step completed.
Attachments (3)
- Figure1.png (349.2 KB) - added by 14 years ago.
- Figure2.png (311.5 KB) - added by 14 years ago.
- Figure3.png (224.0 KB) - added by 14 years ago.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip