Methodology
Pipeline design
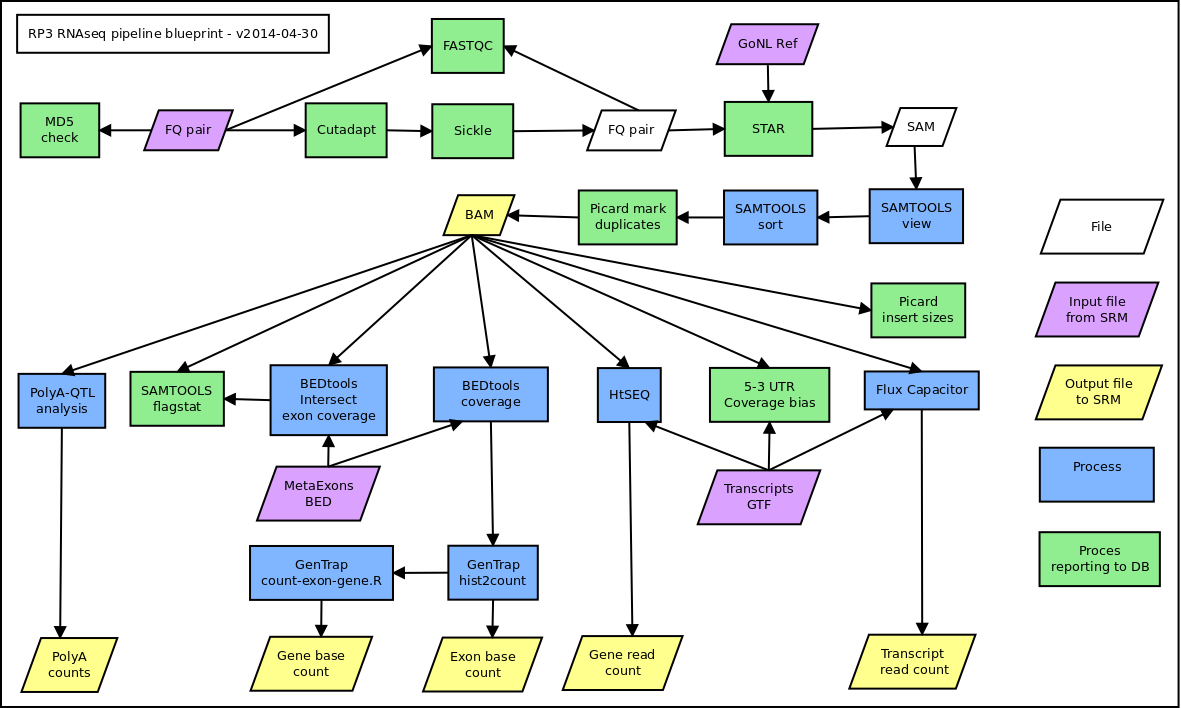
See the attachment (at the pipeline page) for a graphical representation of the pipeline. The LUMC gitlab repository which holds the framework for the pipeline can be found via:
https://git.lumc.nl/groups/rp3
A note on Exon/Gene Quantification using LUMC method
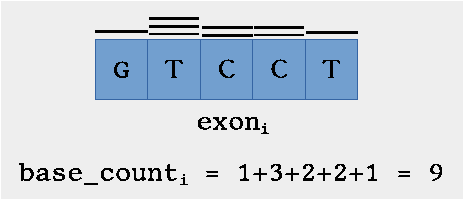
The first step is creating counts for each (meta-)exon (meta exons are identified as overlapping exons) in the (meta-exon) annotation file using bedtools coveragebed (coverageBed) with the BAM file as input. The resulting histogram is converted using the hist2count script which simply counts all the bases for the intervals specified in the hist file (which is the same as the BED file). In the hist file, for a given interval (one single BED record), coverageBed (aka bedtools coverage) outputs the counts for each number of bases. For example, how many bases have 1 coverage, how many have 2, etc. The script simply aggregates this, so that the resulting count file contains the total base counts for each interval.
To quantify genes, the region data is joined based on exons associated to the same individual gene only (Rscript). Some exons are associated to multiple genes. These are separately annotated as meta-exons since (with this method) it's not ascertainable which gene has contributed the data. These meta-features are similarly translated to meta-genes. All the results are written to a file which reports the base-count per (meta)gene.
This situation:
| Gene | -----BBBBBBBBB-
|
| Gene | -AAAAA---------
|
| Exon | -1-2-3-4-5---6-
|
| Coverage | -8-8-9-9-7---7-
|
Results in:
| Gene | Exon | Base count (total coverage) |
|---|---|---|
| A | 1,2 | (8+8) |
| AB | 3 | 9 |
| B | 4,5,6 | (9+7+7) |
Example

Graphical example of how base counts are calculated. (Courtesy of V. Takhaveev)
Hist2count output (*.exon.count)
- chromosome
- start position of the feature (exon)
- end position of the feature (exon)
- total base counts mapped to the feature
- normalized base counts for the region (column 4 divided by (column 3 - (column 2 + 1) ) )
- gene name
Tools and versions
All the required tools for the pipeline are available in a tarball on the grid called toolbox.tar.gz. This tarball is also directly used when the pipeline is running on the grid. The versions of the tools can be found when running, the options we supply are to be found inside the pipeline (align.py). The tarball can be found via:
- toolbox.tar.gz
- srm://srm.grid.sara.nl/pnfs/grid.sara.nl/data/bbmri.nl/RP3/mgalen/toolbox.tar.gz
Combining count files
Do this is steps if there is too many files. First for all Aflowcells then Bflowcells and merge.
For transcript counts:
(echo -n $'\t'; for i in B*.count; do echo ${i%.transcript.count}; done | paste -s; bash -c "paste <(cut -d'\"' -f 2 $(ls B*.count | head -1)) $(for i in B*.count; do echo -n " <(cut -d' ' -f 8 $i | sed 's/;//' )"; done)") > matrixB
For gene counts:
(echo -n $'\t'; for i in B*.count; do echo ${i%.gene.count}; done | paste -s; bash -c "paste <(tail -n +2 $(ls B*.count | head -1) | cut -f 1 ) $(for i in B*.count; do echo -n " <(tail -n +2 $i | cut -f 2)"; done)") > matrixB
For exon counts:
(echo -n $'\t'; for i in B*.count; do echo ${i%.exon.base.count}; done | paste -s; bash -c "paste <(cut -f 1-3 $(ls B*.count | head -1) | tr '\t' _ | sed 's/^/chr_/') $(for i in B*.count; do echo -n " <(cut -f 4 $i)"; done)") > matrixB
See this script for converting exon counts to exon ratios.
Attachments (5)
- base_count_example.png (2.1 KB) - added by 8 years ago.
- pipelineDesignRP3_20130711.pdf (31.2 KB) - added by 8 years ago.
- pipelineDesignRP3_20141030.odp (49.1 KB) - added by 8 years ago.
- pipelineDesignV1404.png (114.1 KB) - added by 8 years ago.
- RNA seq QC 5-2-2016.pdf (1.5 MB) - added by 8 years ago.
{kind=link}
{kind=link}
{kind=link}
Download all attachments as: .zip